Application tools
From HP-SEE Wiki
Contents |
AMBER
Section contributed by IICT-BAS
After testing the performance of modules SANDER and PMEMD of AMBER v.11 with respect to the number of processors and nodes requested, using as a test case cyclodextrin with 100 glucose units (CD100; 2100 CD-atoms and 37459 water molecules; 2.0 ns molecular dynamics simulation; total steps 1000000), it is proceeded further by executing molecular dynamics conformational search with duration 60.0 ns. It is found as an optimum to use module PMEMD with 64 processors (8 nodes with 8 (of 16) processors per node). It is basically well known that the PMEMD module should be used when the use case allows for it, since it is more scalable. In principle using more CPUs is beneficial with respect to the total running time, but the efficiency drops,
GAUSSIAN
Section contributed by IPB-FCUB
Gaussian represent a suite of programs for calculation of electronic structure properties of materials, and is used by chemists, chemical engineers, biochemists, physicists and other scientists. Starting from the fundamental laws of quantum mechanics, Gaussian enables ab-initio calculation of energies, molecular structures, vibrational frequencies and other properties of molecules and chemical reactions in a wide variety of chemical environments. General description of the features of Gaussian is available at the official web site (www.gaussian.com) and at the HP-SEE Wiki Software Stack and Technology Watch / Libraries section (http://hpseewiki.ipb.ac.rs/index.php/GAUSSIAN).
Gaussian03 is ported to the PARADOX 32-bit cluster at the Institute of Physics Belgrade using Linda 7.1 (Gaussian native unit for the parallel processing). Typical usage case-studies in CompChem (http://wiki.hp-see.eu/index.php/CompChem) application are the following:
- Study of mechanisms of chemical reactions: in the reaction of the cylohexanone with the bromoform, two alternative mechanistic pathways are examined.
- Full geometry optimizations on the high level of theoaty, typical DFT or MP2, or the single-point calculations on the geometries obtained on the semi-empirical level of theory, for the diverse set of compounds were performed, aimed to extract molecular descriptors suitable for the linear-free energy relationships (applied in part in Tetrahedron Letters 53 (2012) 553), or for the three-dimensional quantitative structure-activity relationships.
Efficient scaling at PARADOX cluster was obtained using 4 nodes.
MMTSB
Section contributed by IFIN-HH
The MMTSB toolset is a script-language application that provides programs for multiscale sampling modeling and a framework for the development of new applications which require multiple classical all-atom simulation packages for proteins and nucleic acids.
In the HP-SEE infrastructure, MMTSB is currently used by the ISyMAB application, which is hosted on the IFIN_Bio and IFIN_BC clusters, for the analysis and processing of the protein data bank files (PDB), in the framework of the modeling of large biomolecular systems by means of the parallel molecular dynamics NAMD code.
The access to the MMTSB toolset requires the prior authentication through username / password, or SSL certificate, on the ISyMAB web server.
To work with MMTSB, one must first transfer a PDB file on the web server. For this, a couple of options is provided:
a) Uploading from the personal computer
To upload a file to the ISyMAB web server, one must click on the Files icon, and then hit Browse, select the desired file and afther than use the Upload button. The option to "Overwrite existing file" is also available.
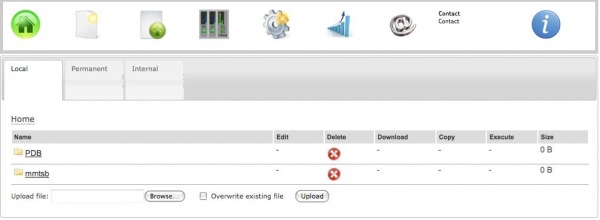
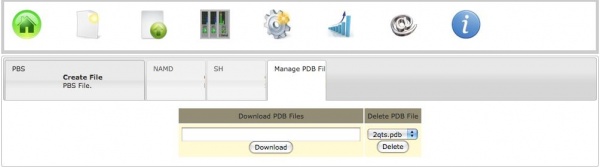
b) Download from RCSB
To download a PDB file directly from RCSB one need to click on the Create Files icon, and then click on the Manage PDB files tab. Here one must enter the code of the structure to be downloaded and then hit Download.
Once the PDB file is available, we can use MMTSB tools to work with it. For instance, in the command-line mode, the extraction of the first N residues from input.pdb and the conversion of the PDB file to the CHARMM format would read:
convpdb.pl -sel 1:N -segnames -out charmm22 -cleanaux input.pdb > output.pdb
MMTSB provides very many commands and associated options as above, which often makes the usage of the command-line mode difficult.
Using the ISyMAB’s GUI considerably reduces the user’s effort in performing these tasks. For this, one must click on the MMTSB Tools icon, which will show the 6 color-coded categories of tools, as shown below:

Each tool has a similar format. Referring to the same example above, from the Structure Preparation category we can choose the convpdb tool and then hit the button under it. This will show the available options that can be used with the convpdb tool.

In the Options text-box one must provide the arguments for the convpdb command, which are described under it. The next thing is to select the Input PDB file to work with, to specify the Output PDB file (filename with extension), and then hit GO.
The output file will be saved in the MMTSB directory, and with the file manager one can download it from ISyMAB web server to the personal computer of the user.
NAMD
Section contributed by IPB-FCUB
NAMD is a parallel classical molecular dynamics code designed for high-performance simulation of large biomolecular systems. Based on charm++ parallel objects, NAMD scales to hundreds of processors on high-end parallel platforms and tens of processors on commodity clusters using gigabit Ethernet. General description of NAMD features are available at the official web page (http://www.ks.uiuc.edu/Research/namd/) and at the HP-SEE Wiki Software Stack and Technology Watch / Library (http://hpseewiki.ipb.ac.rs/index.php/Libraries) section.
Currently, NAMD-2.6 and NAMD-2.8 (including betas) versions are ported on the PARADOX cluster. Both versions are compiled under Charmrun. CompChem (http://wiki.hp-see.eu/index.php/CompChem) application uses NAMD for Molecular dynamics simulation of:
- Sets of small biologically active molecules in isotropic or anisotropic explicit solvents, typically during 20-30 ns. Biasing forced (adaptive biasing force) was used in majority of simulations to obtain free-energy landscapes. Analysis of results connects dynamical behavior of molecules with their selectivity (tumor vs. healthy cells). Two relatively novel concepts, as described in Drug Discov. Today 13 (2008) 285, and J. Chem. Theory Comput. 6 (2010) 35, were merged.
- Simulations similar as described above were performed, using implicit solvent model, aimed to obtain conformations of flexible molecules suitable for linear-free energy relationships. Unique concept so far, Tetrahedron Letters 53 (2012) 553.
- Dynamical behavior of novel fruit allergen isolated in our laboratories, uninhibited, or inhibited were examined. Typically during 5-10 ns, using counterions and explicit solvation with large solvent cluster to simulate experimental conditions (mainly ionic strength). Different ionization states of protein, under different pH values as applied in experiments, is also considered. Those simulations aim to explain experimental observations: different electrophoretic mobility of inhibited and uninhibited protein, as well as difference of proteolytic digestion between the inhibited and the uninhibited protein. Article in preparation.
- Long, typically 60-80 ns, unbiased simulations of ‘approach and entrance’ of acetylcholine esterase dual, non-covalent inhibitors, to enzyme active site. Explicit solvation by large solvent cluster was considered in the each simulation. Set of 10 compounds was simulated so far. Aim of the study is to improve potency of compounds. Such compounds should cure symptoms of the Alzheimer disease. So far one communication submitted.
- Steered molecular dynamics, or unbiased molecular dynamics simulation (typically 10 ns) for the design of the peptidomimetics that should act as inhibitors of the protein-protein interactions. Such compounds should exert activity toward targets involved in life-threatening diseases. Article in preparation.
- Minimization and the equilibration of the few large solvent clusters of the different composition, aimed to be used in standard simulations.
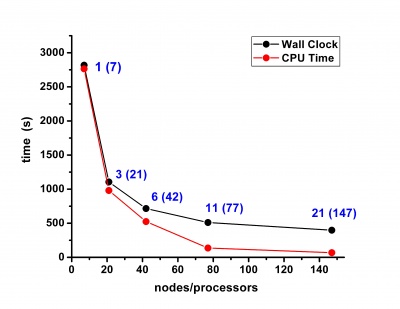
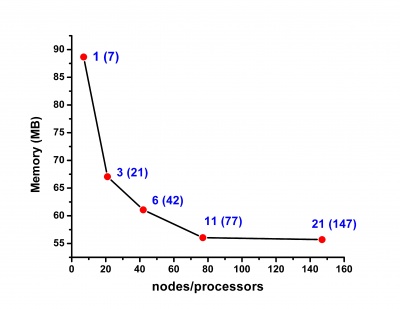
Scalability results of NAMD 2.8 on the PARADOX cluster, measured using the native NAMD benchmark (http://www.ks.uiuc.edu/research/namd/, 10000 steps, 92,224 atoms, 12 Å cutoff + PME every 4 steps, periodic boundary conditions), are given on the figures below.