PCACIC
From HP-SEE Wiki
General Information
- Application's name: Principal component analysis of the conformational interconversions in large-ring cyclodextrins
- Application's acronym: PCACIC
- Virtual Research Community : Computational Chemistry Applications
- Scientific contact : Petko Ivanov, ivanov@bas.bg
- Technical contact : Petko Ivanov, ivanov@bas.bg
- Developers : Petko Ivanov, Institute of Organic Chemistry with Centre of Phytochemistry, Bulgarian Academy of Sciences, Lab. Physical Organic and Computational Chemistry
- Web site: http://wiki.hp-see.eu/index.php/PCACIC
Short Description
A new class of compounds, the large-ring cyclodextrins (LR-CDs), attracted attention in recent years, and advances were marked in the study of their physicochemical properties, in spite of existing difficulties in their synthesis, isolation and purification. Since CDs consist of optically active D-glucose units, they form a pair of diastereoisomeric complexes, usually of different stability, with a racemic compound. There is, however, limited information about either the structure of the macrocycles in solution, or their complex-forming properties.
In view of the difficulties in the purification of individual large-ring cyclodextrins, molecular dynamics simulation techniques provide a useful tool to gain insight into their conformational dynamics and the complex-forming ability. Using molecular dynamics simulations as a conformational search protocol, post-processing of the simulation trajectories is carried out by: (i) the MM/GBSA (Generalized Born/Surface Area (LCPO)) methodology in order to estimate energy data, and (ii) principal component analysis (PCA), also called quasiharmonic analysis or essential dynamics method.
These studies require enormous computational resources. It took only about five years in order to access an order of magnitude longer duration of the simulations for these systems. This is the reason why the use of HPC is necessary.
Previous releases of this application have been run on HPC resources of "Mare Nostrum" at the Barcelona Supercomputing Center.
Problems Solved
The LR-CDs, as the native CDs, are potential reagents for chiral resolution. With this application we can monitor the concerted motions of the atoms of the molecule in a few dimensions, making it easier to visualize and investigate these motions. Our efforts will be devoted to execute additional 50.0 ns simulations for the series CDn (n=24,25,..30) as a test for the convergence of the results from the PCA analysis, as well as treating problems with much higher dimensionality, e.g. CD100.
Scientific and Social Impact
The results will provide invaluable guidelines for the conformational analysis of large rings that present interest as host systems in supramolecular functional architectures.
The results of this work have potential useful applications for design of new materials and in new (nano) technologies, as well as in separation science (molecular recognition and separation of closely related compounds, including geometrical and structural isomers) and for the pharmaceutical industry (different pharmacological activity of enantiomers of a chiral compound).
Collaborations
- UAB-Barcelona
Beneficiaries
- Researchers from computational chemistry community
Number of users
5 number of users: access to HPCG
Development Plan
- Concept: The concept was done before the project started.
- Start of alpha stage: M1 - M4
- Start of beta stage: M4 – M6
- Start of testing stage: M6 – M8
- Start of deployment stage: M9 – M11
- Start of production stage: M12
Resource Requirements
- Number of cores required for a single run: From Tobefilledin to 1000
- Minimum RAM/core required : 1 GB
- Storage space during a single run : < 1TB
- Long-term data storage : < 500 GB
- Total core hours required: 50 000
Technical features and HP-SEE implementation
- Primary programming language : FORTRAN
- Parallel programming paradigm : MPI
- Main parallel code : AMBER
- Pre/post processing code : Own development
- Application tools and libraries: .
Usage Example
A problem in the focus for consideration in our studies is the molecular dynamics (MD) of the conformational interconversions in large-ring cyclodextrins (LR-CDs). Principal component analysis (PCA) was applied for post-processing of trajectories from conformational search, based on MD simulations in aqueous solution (Glycam04 force field and TIP4P water model), with the purpose to elucidate the conformations of some LR-CDs, CDn (n = 10 to 30). The results are in a support for the domination of representative preferred conformations with a specific shape of the LR-CDs for different ranges for the degree of polymerization.1-4
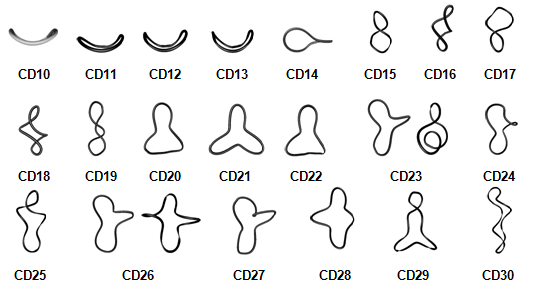 Figure 1. Representative averaged geometries of some LR-CDs.
Figure 1. Representative averaged geometries of some LR-CDs.

With increasing the number of residues, open bent boat-like macrorings (CD10 to CD13) acquire the shapes of twisted eight with two pseudocavities with the size of α-CD and -CD (CD14, CD15), two winded single helical strands, opposing each other at different directions (CD16 to CD19), circularized three-turn single helical structure (CD20, CD21), CD21- and CD26-like conformations (CD22 to CD28), and arbitrary shapes with multiple small cavities (CDn, n>28). Such a classification could assist further experimental research on this new class of compounds. Among the myriad of applications, the CDs are considered also as potential reversible transporters of therapeutic compounds into and out of the cell. Our most recent studies are directed along the examination with molecular dynamics of the conformational behavior of much larger LR-CDs (‘giant cyclodextrins’) – CDn (n=40, 55, 70, 85, 100). The dimensionality of the problem increases drastically with increasing the number of the glucopyranose residues and the number of water molecules. Results from simulations with explicit solvent molecules are sensitive to many parameters (arguments) used in the simulation protocol. Preliminary studies carried out in the past were indicative for possible dependence from the thickness of the water layer (the solvent buffer parameter). We used now 15.0 Å for the solvent buffer parameter. This resulted at significant increase in the dimensions of the periodic TIP4P cubic boxes and the number of water molecules: CD40 (75.9 Å; 13094), CD55 (90.8 Å; 22713), CD70 (93.9 Å; 24982), CD85 (98.5 Å; 28846), CD100 (107.1 Å; 37459). Such studies became feasible for us only after the access to the powerful computational resources of the MareNostrum super-cluster in Barcelona (HPC-BSC, HPC-EUROPA2 project (project number: 228398)), cluster MADARA (the National Science Fund, contract No. ДОО02-52/ RNF01/0110 and contract No. ДОО02-166/TK01/167; http://madara.orgchm.bas.bg) and cluster HPCG at the Institute of Information and Communication Technologies (HP-SEE project, contract No. 261499, EC-FP7). After testing the performance of modules SANDER and PMEMD of AMBER v.11 using as a test case CD100 it was found as an optimum to use module PMEMD with 64 processors (8 nodes with 8 (of 16) processors per node). 100.0 ns simulations were completed for all five LR-CDs and analyses of the simulation trajectories were initiated.

Figure 2. Starting geometries for the simulations. Figure 3. Average structures derived from the PCA analyses of the simulation trajectories, decomposed into 10.0 ns simulation periods (e.g. CD70, from 50.0 ns to 60.0 ns), and compared with the average structure derived from longer simulation (e.g. CD70av (1-60))


Infrastructure Usage
- Home system: HPCG
- Applied for access on: 10.2010
- Access granted on: 10.2010
- Achieved scalability: 64 cores
- Accessed production systems:
- Currently applied for access to other HPC centers in HP-SEE.
- Applied for access on: .
- Access granted on: .
- Achieved scalability: .
- Porting activities: Such computations were previously done by the developers’ team on supercomputing resources in Barcelona and also using their local workstations. In both cases the scalability of the codes for the input data at hand was not very good and lead to exceptionally high running times for each computation. Previously the application has attempted to use also Grid resources, but again the very high running times lead to the necessity to split the computations, leading to loss of precision and extremely cumbersome post-processing. The administrative limits at the home clusters were adjusted to make sure that these problems do not arise in our case. Standing reservations were provided to the developers when they started the production usage, allowing for 8 nodes to be available to them at all times.
- Scalability studies: A good parallel efficiency is optained up to 64 cores on HPCG cluster.
Running on Several HP-SEE Centres
- Benchmarking activities and results:
TABLE - Test for the SANDER and PMEMD modules of AMBER v.11 - cyclodextrin with 100 glucose units (CD100) (2100 CD-atoms and 37459 water molecules; 2.0 ns MD simulation; total steps 1000000)
| No. of Processors | Average Timings for All Steps | Nodes | ppn | -npernode | |||
| Elapsed (s) | Per Step (ms) | ns/day | seconds/ns | ||||
| I. SANDER | |||||||
| 4 | 541836.0 | 541.8 | 0.3 | 270918.0 | 1 | 16 | 4 |
| 8 | 319139.6 | 319.1 | 0.5 | 159569.8 | 1 | 16 | 8 |
| 16 | 324681.0 | 324.7 | 0.5 | 162340.5 | 1 | 16 | 16 |
| 32 | 248933.7 | 248.9 | 0.7 | 124466.8 | 2 | 16 | 16 |
| 64 | 229088.8 | 229.1 | 0.8 | 114544.4 | 4 | 16 | 16 |
| II. PMEMD | |||||||
| 4 | 393667.0 | 393.7 | 0.4 | 196833.5 | 1 | 16 | 4 |
| 8 | 220384.2 | 220.4 | 0.8 | 110219.6 | 1 | 16 | 8 |
| 16 | 177523.6 | 177.6 | 1.0 | 88784.0 | 1 | 16 | 16 |
| 32 | 98923.3 | 98.9 | 1.7 | 49462.0 | 2 | 16 | 16 |
| 64 | 59869.1 | 59.9 | 2.9 | 29942.0 | 4 | 16 | 16 |
| 44868.3 | 44.9 | 3.8 | 22462.2 | 8 | 16 | 8 | |
| 128 | 55923.5 | 56.0 | 3.1 | 27982.7 | 8 | 16 | 16 |
| 37865.1 | 37.9 | 4.6 | 18951.5 | 16 | 16 | 8 | |
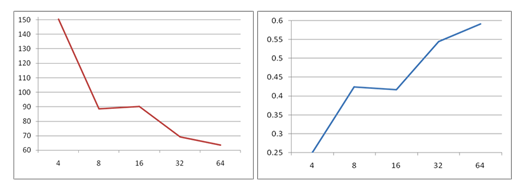
Graphic 1. SANDER scalability and speedup Graphic 2. PMEMD scalability and speedup


The benchmarking calculations for cyclodextrin with 100 glucose units (CD100) showed that using PMEMD one can achieve better scalability than with SANDER. Thus all future computations are performer with PMEMD using 8 nodes with 8 cores per node ( no hyper threading) since this configuration appears to be optimal in this case.
- Other issues: no
Achieved Results
After testing the performance of modules SANDER and PMEMD of AMBER v.11 with respect to the number of processors and nodes requested, using as a test case cyclodextrin with 100 glucose units (CD100; 2100 CD-atoms and 37459 water molecules; 2.0 ns molecular dynamics simulation; total steps 1000000), we proceeded further by executing molecular dynamics conformational search with duration 50.0 ns. We found as an optimum to use module PMEMD with 64 processors (8 nodes with 8 (of 16) processors per node).
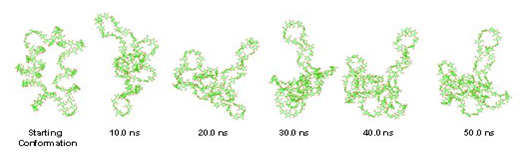 Figure 4. Snapshots of geometries of the macroring conformation of CD100 at different stages of the conformational search.
Figure 4. Snapshots of geometries of the macroring conformation of CD100 at different stages of the conformational search.

Publications
- Ivanov, P. Current Physical Chemistry: Biomolecular Simulations and Applications (a review), in the press.
- P. Ivanov, Conformations of some lower-size large-ring cyclodextrins derived from conformational search with molecular dynamics and principal component analysis, Journal of Molecular Structure, Vol. 1009, 2012, 3–10.
Presentations
- Emanouil Atanassov, Distributed infrastructure for high-performance computing in computational chemistry, Closing Conference of MADARA project, Earth and Man National Museum, 20-21 October 2011, Sofia.
- Emanouil Atanassov, Efficient parallel simulations of large-ring cyclodextrins on HPC cluster, HP-SEE User Forum, 17-19 October 2012, Belgrade.
Foreseen Activities
- to complete the total of 100.0 ns molecular dynamics conformational search;
- additional 10.0 ns simulations will be executed with the purpose to test for the convergence of the conformational search;
- analyses (MM/GBSA; PCA) of the simulation trajectory files with the purpose to obtain energy and structural data, as well as information for the modes of conformational deformations and interconversions. Preparation of the results for publication.