DeepAligner
From HP-SEE Wiki
General Information
- Application's name: Deep sequencing for short fragment alignment
- Application's acronym: DeepAligner
- Virtual Research Community: Life Sciences
- Scientific contact: Miklos Kozlovszky, Gergely Windisch ; m.kozlovszky at sztaki.hu
- Technical contact: Miklos Kozlovszky, Gergely Windisch; m.kozlovszky at sztaki.hu
- Developers: Gergely Windisch, Biotech Group, Obuda University – John von Neumann Faculty of Informatics
- Web site:
http://ls-hpsee.nik.uni-obuda.hu:8080/liferay-portal-6.0.5 http://ls-hpsee.nik.uni-obuda.hu
Short Description
Mapping short fragment reads to open-access eukaryotic genomes is solvable by BLAST and BWA and other sequence alignment tools - BLAST is one of the most frequently used tool in bioinformatics and BWA is a relative new fast light-weighted tool that aligns short sequences. Local installations of these algorithms are typically not able to handle such problem size therefore the procedure runs slowly, while web based implementations cannot accept high number of queries. SEE-HPC infrastructure allows accessing massively parallel architectures and the sequence alignment code is distributed free for academia. Due to the response time and service reliability requirements grid can not be an option for the DeepAligner application.
Problems Solved
The recently used deep sequencing techniques present a new data processing challenge: mapping short fragment reads to open-access eukaryotic (animal: focusing on mouse and rat) genomes at the scale of several hundred thousands.
Scientific and Social Impact
The aim of the task is threefold, the first task is to port the BLAST/BWA algorithms to the massively parallel HP-SEE infrastructure create a BLAST/BWA service, which is capable to serve the short fragment sequence alignment demand of the regional bioinformatics communities, to do sequence analysis with high throughput short fragment sequence alignments against the eukaryotic genomes to search for regulatory mechanisms controlled by short fragments.
Collaborations
Ongoing collaborations so far: Hungarian Bioinformatics Association, Semmelweis University Planned collaboration with the MoSGrid consortium (D-GRID based project, Germany)
Beneficiaries
Serve the short fragment sequence alignment demand of the regional bioinformatics communities. People who are interested in using short fragment alignments will greatly benefit from the availability of this service. The service will be freely available to the LS community. We estimate that a number of 5-15 scientific groups worldwide will use our service.
Number of users
10
Development Plan
- Concept: Done before the project started.
- Start of alpha stage: Done before the project started.
- Start of beta stage: M9
- Start of testing stage: M13
- Start of deployment stage: M16
- Start of production stage: M18
Resource Requirements
- Number of cores required for a single run: 128-256
- Minimum RAM/core required: 4-8 Gb
- Storage space during a single run: 2-5 GB
- Long-term data storage: 1-2 TB
- Total core hours required: 1 500 000
Technical Features and HP-SEE Implementation
- Primary programming language: C,C++
- Parallel programming paradigm: Master-slave, MPI, + Multiple serial jobs (data-splitting, parametric studies)
- Main parallel code: WS-PGRADE/gUSE and C/C++
- Pre/post processing code: BASH script (in-house development)
- Application tools and libraries: BASH script / mpiBLAST (in-house development)
Usage Example
1. HP-SEE’S BIOINFORMATICS ESCIENCE GATEWAY
The Bioinformatics eScience Gateway based on gUSE and operates within the Life Science VO of the HP-SEE infrastructure. It provides unified GUI of different bioinformatics applications (such as BLAST, BWA, or gene mapper applications) and enables end-user access indirectly to some open European bioinformatics databases. gUSE is basically a virtualization environment providing large set of high-level DCI services by which interoperation among classical service and desktop grids, clouds and clusters, unique web services and user communities can be achieved in a scalable way. gUSE has a graphical user interface, which is called WS-PGRADE. All part of gUSE is implemented as a set of Web services. WS-PGRADE uses the client APIs of gUSE services to turn user requests into sequences of gUSE specific Web service calls. Our bioinformaticians need application specific portlets to make the usage of the portal more customized for their work. In order to support the development of such application specific UI we have used the Application Specific Module (ASM) API of the gUSE by which such customization can easily and quickly be done. Some other remaining features were included from WS-PGRADE. Our GUI is built up from JSR168 compliant portlets and can be accessed via normal Web browsers (shown in Fig. 1.).
2. IMPLEMENTATION OF THE GENERIC BLAST WORKFLOW
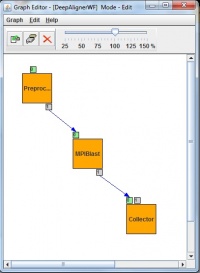
Normal applications need to be firstly ported for use with gUSE/WS-PGRADE. Our used porting methodology includes two main steps: workflow development and user specific web interface development based on gUSE’s ASM (shown in Fig. 2.). gUSE is using a DAG (directed acyclic graph) based workflow concept. In a generic workflow, nodes represent jobs, which are basically batch programs to be executed on one of the DCI’s computing element. Ports represent input/output files the jobs receiving or producing. Arcs between ports represent file transfer operations. gUSE supports Parameter Study type high level parallelization. In the workflow special Generator ports can be used to generate the input files for all parallel jobs automatically while Collector jobs can run after all parallel execution to collect all parallel outputs. During the BLAST porting, we have exploited all the PS capabilities of gUSE.
Parallel job submission into the DCI environment needs to have parameter assignment of the generated parameters. gUSE’s PS workflow components were used to create a DCI-aware parallel BLAST application and realize a complex DCI workflow as a proof of concept. Later on the web-based DCI user interface was created using the Application Specific Module (ASM) of gUSE. On this web GUI, end-users can configure the input parameter like the “e” value or the number of MPI tasks and they can submit the alignment into the DCI environment with arbitrary large parameter fields. During the development of the workflow structure, we have aimed to construct a workflow that will be able to handle the main properties of the parallel BLAST application. To exploit the mechanism of Parameter Study used by gUSE the workflow has developed as a Parameter Study workflow with usage of autogenerator port (second small box around left top box in Fig 5.) and collector job (right bottom box in Fig. 5). The preprocessor job generates a set of input files from some pre-adjusted parameter. Then the second job (middle box in Fig. 5) will be executed as many times as the input files specify. The last job of the workflow is a Collector which is used to collect several files and then process them as a single input. Collectors force delayed job execution until the last file of the input file set to be collected has arrived to the Collector job. The workflow engine computes the expected number of input files at run time. When all the expected inputs arrived to the Collector it starts to process all the incoming inputs files as a single input set. Finally output files will be generated, and will be stored on a Storage Element of the DCI shown as little box around the Collector in.
Due to the strict HPC security constraints, end users should posses valid certificate to utilize the HP-SEE Bioinformatics eScience Gateway. Users can utilize seamlessly the developed workflows on ARC based infrastructure (like the NIIF’s Hungarian supercomputing infrastructure) or on gLite/EMI based infrastructure (Service Grids like SEE-GRID-SCI, or SHIWA). After login, the users should create their own workflow based application instances, which are derived from pre-developed and well-tested workflows.
Infrastructure Usage
- Home system: OE cluster/HU
- Applied for access on: 08.2010
- Access granted on: 08.2010
- Achieved scalability: 4 nodes 8 cores
- Accessed production systems:
NIIF's infrastructure/HU
- Applied for access on: 09.2010
- Access granted on: 10.2010
- Achieved scalability: 96 cores
- Porting activities: The application has been successfully ported,the core workflow was successfully created, the GUI portlet was designed and created.
- Scalability studies: Tests on 32, 59 and 96 cores
Running on Several HP-SEE Centres
- Benchmarking activities and results: At initial phase the application was benchmarkedand optimized on the OE's cluster. After successfull deployment on 32 cores benchmaring was initiated for 59 and 96 cores
- Other issues: There were painful authentication problems and access issues with the supercomputing infrastructure's local storage during porting. Some input parameter assignment optimisation and further study for higher scaling is still required.
Scalability
Benchmark dataset The blast database size was 5.1 GB, and the input sequence size was 29.13 kB. Each measurement was executed 10 times, the average of the 10 executions was taken as the final result Hardware platforms A number of hardware platforms have been used for the testing of the applications. The portlet we have developed is connected to all these different HPC infrastructures and it is the job of the middleware to choose the appropriate for each execution. For our benchmarks we specified the infrastructure the application was supposed to use. The benchmarks were executed on five different HPC infrastructures:
- Debrecen
- Intel Xeon X5680 (Westmere EP) 6 core nodes, SGI Altix ICE8400EX
- 1536 CPU cores
- 6 TB memory
- 0.5 PB storage
- Total capacity: ~18 TFlops
- Budapest (NIIF)
- fat-node cluster using CP4000BL blade
- AMD Opteron 6174 CPUs, 12 cores (Magny Cours)
- ~700 cores
- Total Capacity ~5 TFlops
- Pecs
- SGI UltraViolet 1000 - SMP (ccNUMA)
- CPU: Intel Xeon X7542 (Nehalem EX) - 6 cores
- 1152 cores
- 6 TB memory
- 0.5 PB memory
- Total capacity: ~10 TFlops
- Szeged
- fat-node cluster using CP4000BL blade
- AMD Opteron 6174 CPUs, 12 cores (Magny Cours)
- 2112 cores
- 5.6 TB memory
- 0.25 PB storage
- Total Capacity ~14 TFlops
- Bulgaria
- Blue Gene/P with PowerPC CPUs
- 2048 PowerPC 450 based compute nodes
- 8192 cores
- 4 TB memory
Software platforms The applications were tested using multiple software stack
- Different MPI implementations
- openmpi_gcc-1.4.3
- openmpi_open64-1.6
- mpt-2.04
- openmpi-1.4.2
- openmpi-1.3.2
- Different compilers
- opencc
- icc
- openmpi-gcc
Each of the different hardware platforms have multiple MPI environments. We have tested our applications with multiple versions. There usually is one specific preferred at each of the HPC centers which we preferred using.
- Execution times
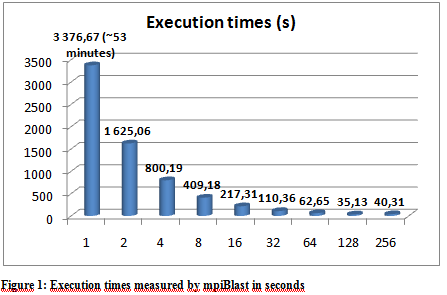
The following graphs show the results of the executions. The execution times varied a little depending on the hpc ceter used, but they were more or less stable so we only include the results from the Budapest server. The following graphs show the result of multiple executions of mpiBlast on the same database with the same input sequence on the same computer. The only difference being the number of CPU cores allocated to the MPI job . Figure 1 shows the execution times measured by mpiBlast. If executed on just one CPU it takes 3376 seconds for the job to finish (about 53 minutes). As we can see the applications scales well, the execution times drop when we add more and more CPUs. The scalability is linear until 128 cores.
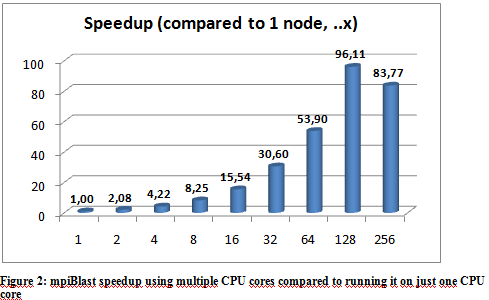
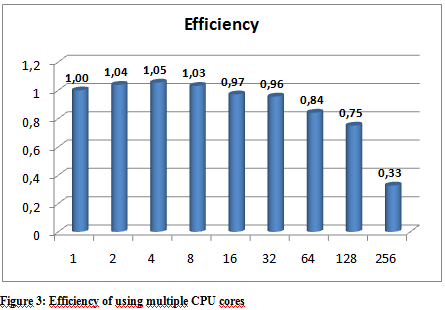
Further optimization The first task when using mpiBlast is to split the blast database into multiple fragments. According to previous research, the number of database fragments have a direct impact on the performance of the application. Finding an optimal number was essential, so our database was split into different sizes. Figure 4 shows the measured execution times. The measurements were executed on 64 cores. The execution times show that the application performs best when the number of DB segments are integer multiples of the number of CPU cores. The reason is straightforward: this is the only way an even data distribution can be achieved amongst the cores.
- Profiling
The two applications we have created share some of the code base which results in a similar behavior. Both applications consist of three jobs in a WS-PGrade workflow with job 1 being the preprocessor, job 2 doing the calculations and job 3 collecting the results and providing it to the user. The current implementation for the preprocessing is serial, we have investigated parallelizing but according to our profiling approximately 0.02 % of the total execution time is spent on Job 1 in Deep Aligner, so yields no real performance gain but can cause problems so we voted againts it. Job3 is 0.01% - most of the work is done in Job2. Job2 consists mainly of mpiBlast, the profiling shows the following results.
Execution time ratio of the jobs in the whole Deep Aligner portlet. Job1: 0,02% Job2: 99,97% Job3: 0,01%
Execution time ratio inside Job2
Init: 1,79%
BLAST: 97,18%
Write: 0,19%
Other: 0,84%
- Memory
Memory usage while executing the application. The results come from the maxvmem parameter of qacct: 1: 1,257 2: 2,112 4: 3,345 8: 4,131 16: 5,434 32: 6,012 48: 4,153 64: 8,745 96: 9,897 128:12,465
As we can see the memory consumtion (measured by qacct) increases as the number of cores is increased.
- Communication
mpiBlast uses a pre-segmented database and each node have their own part where it searches for the input sequence so the communication overhead is very small.
- I/O
I/O as measured using the io parameter of qacct: 1: 0,001 2: 0,001 4: 0,002 8: 0,003 16: 0,004 32: 0,011 48: 0,016 64: 0,019 96: 0,027 128:0,029
As we can see on the previous table the I/O use increases as we increase the number of CPU cores in the job.
- Analysis
From our tests, we conclude that our application scales reasonably well up until about 128 cores. When the appropriate MPI implementation is used on the HPC infrastructure the performance figures are quite similar – the scalability results are within the same region as expected. The number of database fragments play a significant role in the whole application and the best result can be obtained when that number is equal to or is an integer multiple of the number of cores. We have also noted that because of the high utilization of the supercomputing centers real life performance – wall clock time measured from the initialization of the job until the results are provided – could be better when using a smaller number of cores because small jobs tend to get scheduled easier and earlier.
Achieved Results
The DeepAligner application was tested with parallel short DNA sequence searches successfully. So far publications are targeting mainly the porting of the application, publication of more scientific results is planned.
Publications
- G. Windisch, M. Kozlovszky, Á. Balaskó;Performance and scalability evaluation of short fragment sequence alignment applications;HPSEE User Forum 2012
- M. Kozlovszky, G. Windisch, Á. Balaskó;Short fragment sequence alignment on the HP-SEE infrastructure;MIPRO 2012
- M. Kozlovszky, G. Windisch; Supported bioinformatics applications of the HP-SEE project’s infrastructure; Networkshop 2012
Foreseen Activities
Parameter assignements optimisation of the GUI, more scientific publications about short sequence alignment.