CompChem
From HP-SEE Wiki
General Information
- Application's name : Quantum Mechanical, Molecular Mechanics, and Molecular Dynamics computation in chemistry
- Application's acronym: CompChem
- Virtual Research Communities : VRC Computational Chemistry
- Scientific contact : Ivan Juranic, ijuranic@chem.bg.ac.rs
- Technical contact : Ivan Juranic, ijuranic@chem.bg.ac.rs
- Developers : Ivan Juranic, University of Belgrade, Faculty of Chemistry, Republic of Serbia
- Web site : http://www.chem.bg.ac.rs/~ijuranic/prezengl.htm
Short Description
The project deals with modeling of molecular structure and mechanism of chemical reactions. Quantum mechanical, ab-initio, and DFT calculations will be applied for molecules that consist up to 250 atoms. Molecular mechanics and molecular dynamics simulation will be used for examination of ligands interaction with proteins and other biomacromolecules, as well as for conformational sampling of small molecules in explicit solvent cluster. In such simulations size of ligands should be up to 300 atoms; while biomacromolecule can have size of more than ten thousands of atoms. In the present stage, simulations were done by NAMD2.8.
Our research is directed toward:
a) examination of the free-energy landscapes of highly potent and very selective antitumor agents, named CSAB (see Journal of Medicinal chemistry, 48, 5600-5603, (2005); The XVI European Symposium on Quantitative Structure-Activity Relationships and Molecular Modelling, 10-17 September 2006 Mediterranean Sea / Italy. Final program& General Information pp. 274-275; and The 18th European Symposium on Quantitative Structure-Activity Relationships, 19-24 September 2010, Rhodes, Greece. Final Program & Abstract Book, pp. 278-279), in explicit solvent clusters, having different dielectric constants, H-bonding ability, and polarity, by medium lasting (~ 30ns) molecular dynamic simulations coupled with adaptive biasing force calculation;
b) Molecular dynamics simulations (~ 80 ns each) for “approach and entrance” of the group of compounds, which act as dual acetylcholine esterase (AChE) inhibitors, to enzyme active site. Whole systems (enzyme + inhibitor) for each compound are embedded in properly sized (large) solvent cluster. Last stage of each simulation includes adaptive biasing force calculation.
Several components of this approach require massive study of the vast parameter space and include tightly-coupled simulations with large memory requirements. Testing, benchmarking and production runs of some of the components can be done on standard Grid-type Linux clusters, but the core components require low-latency parallel environment. Postprocessing of trajectories, as well as output of (coupled) free-energy calculations, are also often memory demanding, so it is suitable to do such analysis on the nodes associated with the cluster where simulation were performed.
Problems Solved
Both group of compounds described in previous section are designed, synthesized and biologically tested by our group. First group of compounds represent entirely new chemotypes, as well as new chemical entities; while second group comprises new chemical entities.
Simulations described in application description under a) – CSAB – should give insight of selectivity of compounds (potency toward tumors/ toxicity toward healthy cells), which are, according to existing experimental data, highly correlated with molecular flexibility. Also, we try to find most appropriate force field among those that are available, for this type of simulation; and to examine advantages and possible disadvantages of methods of calculation used, which are relatively novel and were not applied on the systems comparable with ours, so far.
Simulations described in application description under b) – interaction of dual AChE inhibitors with the enzyme – should give insight on intramolecular interactions between the enzyme and examined compounds, and should give hint for most favorable structural modifications of compounds in order to improve their potency. To the best of our knowledge similar simulations were not reported in literature so far.
Scientific and Social Impact
Within major applicable goals of scientific knowledge now and in close future, few are priority: novel energy sources, novel materials, and novel medicines. First two goals arise from decreasing amount of fuels originated from natural sources in the World; the third one is ultimate consequence of the rise of population and growing older population in the developed countries. Developing of new medicines in a rational way includes recognition of target (enzyme, protein) that causes a specific physiological disorder, and developing of molecules that could modify those targets in a manner which attenuate symptoms or cure the illness. Looks simple, but in practice is still far reachable goal for treating different kinds of tumors, retroviral infections, or neurodegenerative diseases. Along with this, relatively quick acquired resistance of pathogenic bacteria and viruses to existing marketed drugs made searching for the new drugs the never-ending race. In silico analysis of targets, analysis of interaction of targets with the small molecules that should modulate their physiological outcome, and search for small molecules that could favorably interact with the targets, is the one of the top-priority components of drug design framework. Pharmaceutical companies spend hundreds of millions of dollars per year for R&D projects. Within our group, three classes of promising lead compounds, that targeted human tumors, multidrug resistant bacteria (superbugs) and key enzyme responsible for Alzheimer disease, is developed so far. Within the actual project we aimed to further, and more efficiently use in silico techniques, which so far have a key importance for design of all three classes of molecules, as guidance for additional modification/improvement of structures, in order to obtain more potent and/or more selective compounds within all three examined classes.
Project should result with widening the pool of potentially healing substances (for the concept of chemical space, closely associated with previous statement see Nature 432(432) 823–865 (2004)), i.e. final results should facilitate curing life-threatening diseases or contribute to significant improvement of life quality in chronic illnesses: Non est vivere sed valere vita. Along with this, young researchers will be instructed/educated in using in silico techniques for rational design of bioactive molecules having desired mode of action.
Collaborations
The School of Pharmacy, University of London, GB; and The Chemistry Department, Laboratory of Organic Chemistry, University of Athens, and the National Hellenic Research Foundation, Athens, Greece.
Beneficiaries
Results of simulations developed during this project will be used for design of new compounds within our group (Department of Chemistry-IChTM, University of Belgrade and Faculty of Chemistry, University of Belgrade), as well as in collaborative work with research groups assigned under 'collaborations'
Number of Users
17
Development Plan
- Concept: done before project start
- Start of alpha stage: done before project start
- Start of beta stage: done before project start
- Start of testing stage: M1
- Start of deployment stage: M3
- Start of production stage: M4
Resource Requirements
- Number of cores required for a single run: 8 – 128, probably more
- Minimum RAM/core required: 512 MB
- Storage space during a single run: 5-10 GB
- Long-term data storage: 90 GB
- Total core hours required: 2 000 000
Technical features and HP-SEE implementation
- Primary programming language : Charm++ programs- written in C++ with some library calls and an interface description language for publishing Charm++ object
- Parallel programming paradigm : Hybrid- Charm++\Converse, a machine independent parallel programming system
- Main parallel code : NAMD 2.9, http://www.ks.uiuc.edu/Research/namd/. Parallel codes : Firefly (PC GAMESS) 7.1G, http://classic.chem.msu.su/gran/firefly/index.html; CPMD, http://www.cpmd.org/; ORCA, http://www.mpibac.mpg.de/bac/logins/neese/description.php , Mead 2.2.9 http://www.stjuderesearch.org/site/lab/bashford. Sequential code : AutoDock Vina 1.0 http://vina.scripps.edu/; OMEGA, ROCS, EON, FRED - substituted by OEdocking suite, SZYBKI, SZMAP http://www.eyesopen.com/products. The OpenEye software able to run both in parallel and sequential modes
- Pre/post processing code : Do not exist
- Application tools and libraries: Charm++, OpenMPI, MPICH
Usage Example
1. Mechanism of Boyland-Sims oxidation, see reference 1 bellow
2. Conformational space of the set of bioactive compounds (J. Med. Chem. 2005, 48, 5600) in explicit n-octanol/water, water, dimethylsulfoxide, chloroform, and implicit ethanol was examined during 20-30 ns. Free-energy landscape of examined molecules was mapped using adaptive biasing force calculations (ABF - http://www.edam.uhp-nancy.fr/ABF/applications.html). Rational and the results, in part, can be found in reference 2, see bellow. Some informative figures


3. Molecular dynamics simulation (60 – 80 ns) of entrance of dual AChE inhibitors, made by our group, to enzyme active site

4. Search for conformations of flexible molecules appropriate to be used in Linear Free Energy Relationships. Free-energy landscape of compounds are mapped using adaptive biasing force calculations, followed by semiempirical and DFT calculations; reference # 4.
5. Probing AChE active site flexibility by molecular dynamics and alignment-independent 3D QSAR, merging structure and ligand based drug design. Article in preparation
6. Experimental observations challenged by molecular dynamics, the new fruit allergen. Article in preparation.
Infrastructure Usage
- Home system: PARADOX - IPB
- Applied for access on: 09/2010
- Access granted on: 09/2010
- Achieved scalability: 128 cores
- Scalability studies: NAMD 2.9 - 86_64-ibverbs, CHARMM FF, ~ 87000 atoms, 14 Å cutoff , PME, PBC, SMD (reference, constraints, pulling force), DCD writing every 1000 steps / 50 000 steps
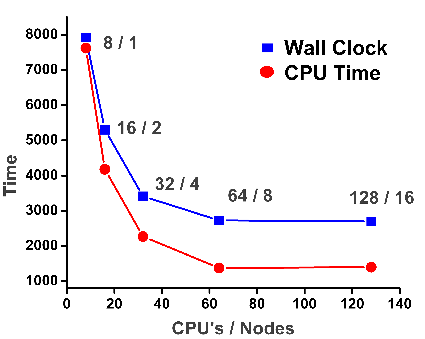

- Accessed production systems:
- HPCG/BG
- Applied for access on: 02/2012
- Access granted on: 03/2012
- Achieved scalability: in progress
- Porting activities: source codes needed already ported
- Scalability studies: NAMD 2.9 - 86_64-ibverbs, CHARMM FF, ~ 87000 atoms, 14 Å cutoff , PME, PBC, SMD (reference, constraints, pulling force), DCD writing every 1000 steps / 50 000 steps
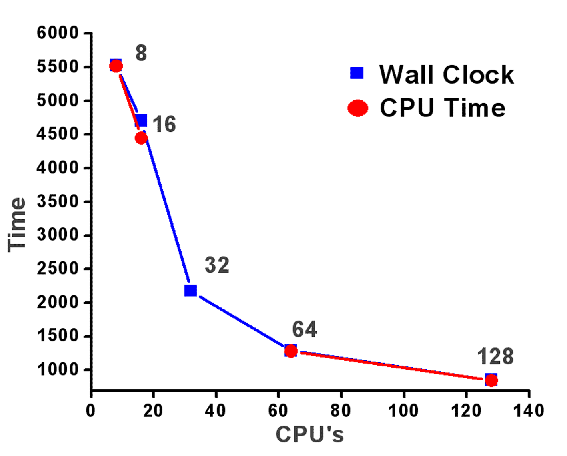

Running on Several HP-SEE Centres
- Benchmarking activities and results: /
- Other issues: Application for the HPCG/BG granted, benchmarking shown above
Achieved Results
See under both 'publications' and 'usage examples', due to policy valid for majority of the scientific journals, the results prepared for publication cannot be publicly shown before acceptance.
Publications and Presentations
1. Budimir Marjanović, Ivan Juranić, and Gordana Ćirić-Marjanović: “Revised Mechanism of Boyland Sims Oxidation”. J. Phys. Chem. A 2011, 115(15), 3536-3550, doi:10.1021/jp111129
2. B J. Drakulić, A Pedrretti, M Zloh, V Slavnić, I O. Juranić, M M. Dabović “Range And Sensitivities of 2-[(Carboxymethyl)sulfanyl]-4-oxo-4-arylbutanoic Acids Property Spaces. Part 2. Multidimensional Free Energy Landscapes” 18th European Symposium on Quantitative Structure – Activity Relationships, “Discovery Informatics & Drug Design”, 19-24 September 2010, Rhodes, Greece. Organized by The Cheminformatics and QSAR society and sponsored by European Federation for Medicinal Chemistry (EFMC).
3. M.D. Vitorović-Todorović, I.N. Cvijetić, I.O. Juranić, B.J. Drakulić “Chemometric Clasification and Structure-Activity Study of Cholinesterase Dual Inhibitors” EuroAnalysis 2011, European Conference of Analytical Chemistry, Belgrade, Serbia, September 2011.
4. B.J. Drakulić, A.D. Marinković, I.O. Juranić "On the choice of optimal conformation in linear free-energy relationships. Reactivity of 2-[(carboxymethyl)sulfanyl]-4-oxo-4-arylbutanoic acids with diphenyldiazomethane" Tetrahedron Lett. 53 (2012) 553–556; doi: 10.1016/j.tetlet.2011.11.097
5. B.J. Drakulić, October 5, 2011 Use of HPC Resources in HP-SEE Project - CompChem in High Performance Computing - HP-SEE Dissemination and Training, Sarajevo http://indico.ipb.ac.rs/getFile.py/access?contribId=4&resId=0&materialId=slides&confId=226
6. B.J. Drakulić, November 28, 2011 NAMD on PARADOX, input preparation, job submission and monitoring http://indico.ipb.ac.rs/conferenceDisplay.py?confId=253
7. B.J. Drakulić, C-Z. Dragos, V. Ionut, P. Ivanov, N. Dodoff, A. Karaivanova, I.O. Juranić ' The medicinal chemistry-related applications in HP-SEE project. An end-user view’ 15th Hellenic Symposium on Medicinal Chemistry, Athens, Greece, May, 2012.
8. M.D. Vitorović-Todorović, B.J. Drakulić ‘Unconstrained, unbiased, MD simulation of the low nanomolar dual AChE inhibitor interaction with the enzyme’ 15th Hellenic Symposium on Medicinal Chemistry, Athens, Greece, May, 2012.
9. Jelena Randjelović ‘Firefly Scientific Software’, in Tuning and Optimization of HPC Application (http://indico.ipb.ac.rs/conferenceDisplay.py?confId=279), Institute of Physics, Belgrade, Serbia, Jun 01, 2012.
10. B. Drakulić ‘OpenEye Scientific Software’, in Tuning and Optimization of HPC Application (http://indico.ipb.ac.rs/conferenceDisplay.py?confId=279), Institute of Physics, Belgrade, Serbia, Jun 01, 2012.
11. B.J. Drakulić, I.O. Juranić 'CompChem (RS) application in the HP-SEE project. Resources, source codes, strengths and weaknesses' 50th Meeting (Golden Jubilee) of the Serbian Chemical Society, June 14-15, 2012, Belgrade, Serbia.
12. M.D. Vitorović-Todorović, C. Koukoulitsa, I.O. Juranić, B.J. Drakulić ‘4-Aryl-4-oxo-N-phenyl-2-aminylbutyramides with interesting AChE/BChE selectivity profile’, 19th EuroQSAR ‘Knowledge enabled ligand design’ Vienna, Austria, 26-30 August 2012
13. B.J. Drakulić, Ž.S. Žižak, T.P. Stanojković, S. Ristić, D.M. Gođevac, I.O. Juranić ‘2-[(Carboxymethyl)sulfanyl]-4-oxo-4-arylbutanoic acids. Part 3. Pharmacology and conformational preferences in different solvents. The NMR/MD study’ 19th EuroQSAR ‘Knowledge enabled ligand design’ Vienna, Austria, 26-30 August 2012
14. Maja D. Vitorović-Todorović, Ilija N. Cvijetić, Ivan O. Juranić, Branko J. Drakulić 'The 3D-QSAR study of 110 diverse, dual binding, acetylcholinesterase inhibitors based on alignment independent descriptors (GRIND-2). The effects of conformation on predictive power and interpretability of the models' Journal of Molecular Graphics and Modelling, 38 (2012) 194-210, doi: 10.1016/j.jmgm.2012.08.001
15. Vesna D. Vitnik, Željko J. Vitnik, Ivan O. Juranić ‘Carbenic vs. ionic mechanistic pathway in reaction of cyclohexanone with bromoform’ Journal of Molecular Modeling, 18 (2012) 4721-4728, doi: 10.1007/s00894-012-1468-2.
16. Ž.J. Vitnik, V.D. Vitnik, S.V. Pokorni, I.O. Juranić ‘Correlation of pKa values for series of benzoic acids with the theoretically calculated atomic charges’ Physical chemistry 2012, 11th International Conference on Fundamental and Applied Aspects of Physical Chemistry, Belgrade, September 2012
17. I.O. Juranić 'Use of high performance computing in chemistry' HP-SEE Marketing Event, Tuzla, October 2012
18. I.O. Juranić 'Use of High Performance Computing in (Bio)Chemistry', invited lecture, HP-SEE User Forum, Belgrade, 19 October 2012
19. B.J. Drakulić, M. Gavrović-Jankulović 'Dynamics of uninhibited and covalently inhibited cysteine protease on non-physiological pH' HP-SEE User Forum, Belgrade, 19 October 2012
20. B.J. Drakulić, I.O. Juranić 'Free-energy surfaces of 2-[(carboxymethyl)sulfanyl]-4-oxo-4-arylbutanoic acids. Molecular dynamics study in explicit solvents' HP-SEE User Forum, Belgrade, 19 October 2012
21. I.N. Cvijetić,I.O. Juranić, A. Pedretti, G. Vistoli, B.J. Drakulić 'In the search of the HDAC-1 inhibitors. The preliminary results of ligand based virtual screening' HP-SEE User Forum, Belgrade, 19 October 2012
22. I.N. Cvijetić, M.D. Vitorović-Todorović, I.O. Juranić, B.J. Drakulić 'Conformational preferences of 2-[(2-hydroxyethyl)sulfanyl]-4-oxo-4-(2,4-diisopropylphenyl)-butanoic acid phenylamide. The NMR/MD study' First International Conference of young chemist of Serbia, Belgrade, 20 October 2012
23. A.Z. Simić, T.Ž. Verbić, M.N. Sentić, M.P. Vojić, I.O. Juranić, D.D. Manojlović 'Study of ellagic acid electro-oxidation mechanism' Monatshefte für Chemie 144 (2012), 121-128, doi:10.1007/s00706-012-0856-8
24. J. Ranđelović, S. Erić, V. Savić 'Computational study and peptide inhibitors design for the CDK9-cyclin T1 complex' Journal of Molecular Modeling 19 (2013) 1711-1715, doi: 10.1007/s00894-012-1735-2
25. M.M. Grozdanovic, B.J. Drakulić, M. Gavrović-Jankulović 'Conformational mobility of active and E-64-inhibited actinidin' Biochimica et Biophysica Acta - General Subjects 1830 (2013), 4790–4799, doi: 10.1016/j.bbagen.2013.06.015
26. I.N. Cvijetić, D.D. Petrović, T.Ž. Verbić, I.O. Juranić, B.J. Drakulić 'Human serum albumin binding of 2-[(carboxymethyl)sulfanyl]-4-oxo-4-arylbutanoic acids' 3rd World Conference on Physico-Chemical Methods in Drug Discovery and Development, 22-26 September 2013, Dubrovnik, Croatia
27. I.N. Cvijetić, M.D. Vitorović-Todorović, I.O. Juranić, B.J. Drakulić 'Reactivity of (E)-4-aryl-4-oxo-2-butenoic acid arylamides toward 2-mercaptoethanol. A LFER study' Monatshefte für Chemie 144 (2013) 1815-1824, doi: 10.1007/s00706-013-1084-6
28. B.J. Drakulić, M.D. Vitorović-Todorović 'Using Simulations Plus’ ADMET Predictor as an Accompanying Tool in QSAR/LFER Modeling: Case Studies' Simulations Plus’ software: its applications in various aspects of pharmacy and chemistry. Seminar, Faculty of Pharmacy, Belgrade, September 20, 2013
29. M.D. Vitorović-Todorović, C. Koukoulitsa, I.O. Juranić, Lj.M. Mandić, B.J. Drakulić 'Structural modifications of 4-aryl-4-oxo-2-aminylbutanamides and their acetyl- and butyrylcholinesterase inhibitory activity. Investigation of AChE-ligand interactions by docking calculations and molecular dynamics simulations' European Journal of Medicinal Chemistry 81 (2014) 158-175, doi: 10.1016/j.ejmech.2014.05.008
30. I.N. Cvijetić, M.D. Vitorović-Todorović, I.O. Juranić, Ð.J. Nakarada, M.D. Milosavljević, B.J. Drakulić 'Reactivity of (E)-4-aryl-4-oxo-2-butenoic acid phenylamides with piperidine and benzylamine: kinetic and theoretical study' Monatshefte fur Chemie 145 (2014) 1297-1306, doi:10.1007/s00706-014-1223-8
31. I.N. Cvijetić, D.D. Petrović, T.Ž. Verbić, I.O. Juranić, B.J. Drakulić 'Human Serum Albumin Binding of 2-[(Carboxymethyl)sulfanyl]-4-oxo-4-(4-tert-butylphenyl)butanoic Acid and its Mono-Me Ester' ADMET & DMPK 2 (2014) 126-142, doi: 10.5599/admet.2.2.28
32. J.M. Marković, N.P. Trišović, D.Mutavdžić, K. Radotić, I.O. Juranić, B.J. Drakulić, A.D. Marinković 'Solvatochromism of symmetrical 2,6-distyrylpyridines. An experimental and theoretical study' Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 135 (2015) 435-446, doi: 10.1016/j.saa.2014.07.023
33. M. Kalinić, M. Zloh, S. Erić 'Structural insights into binding of small molecule inhibitors to Enhancer of Zeste Homolog 2' Journal of Computer-Aided Molecular Design, in press, doi: 10.1007/s10822-014-9788-1
Foreseen Activities
Version 2.9 of NAMD, MD source code, improve in some extent the issue of slow communication between nodes. Still, using the same program on HPCG/BG, with Infiniband communication give significantly better results; see under: Infrastructure Usage/Scalability studies.
Installation of the AutoDock Vina 1.0, as well as OpenEye applications (OMEGA, ROCS, EON, FRED and SZYBKI) allows virtual screening (VS) on large databases of compounds (including structure-based and ligand-based VS). Even those applications do not use more than one-three nodes efficiently; running many sequential jobs in parallel will yield desired results.
Installation of Desmond source code for MD still is the issue, because of many dependencies. We will work in future to solve existing problems.
With installed source codes on existing infrastructure on our home cluster (PARADOX-IPB), calculation on the MM to ab initio levels is possible. So, along modelling tasks related to rational drug design, the examination of the path of chemical reactions, prediction of the spectral properties of compounds and similar tasks are feasible.